
MAPS Reaches The Last Mile: Part I
Last week, The Multidisciplinary Association for Psychedelic Studies (MAPS) published peer-reviewed data from the second Phase III confirmatory trial of MDMA-Assisted Therapy (MDMA-AT) for PTSD in the journal Nature Medicine.
From the paper’s discussion section (emphasis added):
“In this confirmatory phase 3 study of participants with moderate to severe PTSD, MDMA-AT significantly improved PTSD symptoms and functional impairment, as assessed by CAPS-5 and SDS, respectively, compared to placebo with therapy over 18 weeks. Notably, 45 of 52 (86.5%) participants treated with MDMA-AT achieved a clinically meaningful benefit, and 37 of 52 (71.2%) participants no longer met criteria for PTSD by study end.”

For those who have been following psychedelic research for some time, these results were pretty much expected; after all, this is the second of MAPS’ Phase III studies, and the results are similarly positive to those published in 2021.
But for the general public, data like this and the dissemination of the results by mainstream outlets like The New York Times and Oprah are bringing more and more attention and awareness of the therapeutic utility of these culturally maligned compounds.
A key dynamic of the “psychedelic renaissance” is that, with every major scientific and policy development, the attention and awareness of their utility as therapeutics ratchets up.
And with the rise in awareness, the interest in and demand for psychedelic therapeutic experiences increases.
As we covered a few weeks ago, a recent study from the National Institute on Drug Abuse found psychedelic use is at an all-time high.

However, despite this interest and demand, their legal status remains unchanged.
The significance of this study is not just the positive data but rather because it marks the culmination of a 40-year sojourn1 that includes 21 MAPS-sponsored clinical trials of MDMA for PTSD and kicks off the beginning of a process that can change the legal status of MDMA once and for all and bring MDMA-AT to market.
In other words, MAPS has reached the “last mile” of a 40-year odyssey, and the finish line is in sight.
The Last Mile
The concept of the “last mile” comes from the supply chain, travel, and logistics domains.
Let’s say you travel from New York to San Francisco. While you are in flight, you are moving 500 miles per hour. The price-to-distance-to-time ratio is at its maximum. You are covering more ground at less cost in less time.
Once you have landed, the cost-to-time-to-distance equation goes up exponentially. You cover less ground over a longer time at a higher cost. An Uber is slower, less cost-efficient, and less time-efficient than a jet. It takes 30 minutes to travel the ten miles from SFO to the Mission District. In the air, it took 60 seconds to cover 10 miles.
This has become known as the “last mile problem.”
In MAPS’ quest to bring MDMA-AT to those suffering from PTSD, they have reached the last mile of the journey, where things become complex.
Breakthrough Therapy Designation & Special Protocol Assessment
Just because a sponsor has conducted 21 clinical trials, completed Phase III dosing, and published positive data does not guarantee the FDA will approve the drug.
What comes next is the submission of a New Drug Application (NDA), which we’ve heard could happen as soon as this fall.
The NDA is the formal last step taken by a drug sponsor to request that the FDA approve a new drug for sale and marketing in the United States.
The purpose is to demonstrate to the FDA that the drug is safe and effective for its intended use, manufactured correctly, and appropriately labeled and packaged. It will include information about the chemistry and manufacturing processes, results from animal and preclinical toxicology studies, data about how the drug is absorbed, metabolized, and excreted, data from Phase I-III clinical trials, updated safety reports, and plans for pediatric trials, among other things.
A standard review process requires the FDA to come to a decision within ten months of the NDA submission. If an NDA is awarded Priority Review, it means the FDA will make a decision within six months.
Two distinctions can help the review timeline and increase the likelihood of approval.
In 2017, the FDA awarded Breakthrough Therapy Designation to the MDMA-AT program. Breakthrough status allows “All Fast Track” designation features and “intensive guidance on an efficient drug development program, beginning as early as Phase 1.”
MAPS also has a Special Protocol Assessment (SPA) in place with the FDA, which allows drug sponsors to agree on the study design, size, endpoints, and statistical measurements with the FDA.
Although achieving the apriori benchmarks set by a SPA does not guarantee approval, meeting these benchmarks is a very good sign.
Unique Features of MDMA-AT
If the potential regulatory approval of a Schedule I substance wasn’t enough, there are several other factors that need consideration prior to or upon FDA approval, including:
Risk Evaluation and Mitigation Strategies (REMS)
Drug labeling
Staff and oversight needs
Federal rescheduling by the DEA and state-by-state rescheduling
MDMA-AT is a drug and therapy combination product—which I believe is the first time the FDA has had to consider the psychotherapy process in an NDA— and, combined with the long-duration altered state of consciousness, certain safeguards will likely be put in place. These include a REMS and staffing and patient monitoring requirements.
Consider that the study protocols included three preparation sessions, three dosing sessions, and nine integration sessions, and it is clear that this is not merely the approval of another drug that patients pick up from a pharmacy and take on their own.
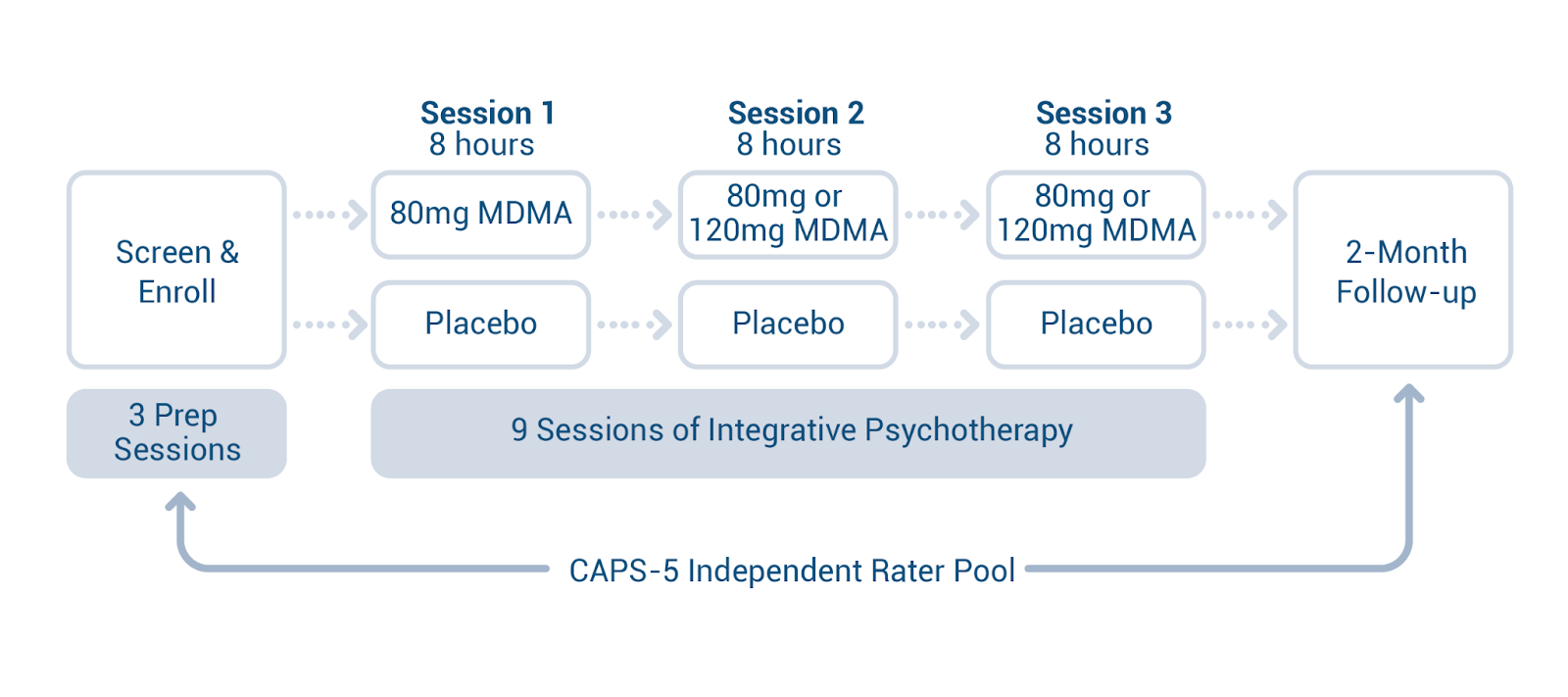
The FDA doesn’t regulate the practice of medicine or therapy, but from this unique situation, we may see guidance on the number and training level of providers required to administer MDMA-AT.
Then, one of the most crucial aspects of this whole process will be the language written on package inserts that accompany the drug, called Drug Labeling, which is a critical component of FDA approval because it communicates the necessary information about the drug to ensure its safe and effective use. It is also key for insurance reimbursement.
And finally, upon FDA approval, the DEA will have 90 days to reschedule MAPS-manufactured MDMA by mandate of the Controlled Substances Act. Once this is done, the commercial route can begin.
All told, with an anticipated NDA submission by the end of the year, a 6-10 month review period, and a 90-day window for the DEA to reschedule, we could see commercially available MDMA-AT within a year from now.
In Part II, we’ll look at the factors that will affect the rollout of MDMA-AT, which includes a sufficient number of trained therapists, clinics equipped for the unique demands, and institutional alignment with, for example, the VA.
Is it a coincidence that Moses spent 40 years in the desert before God raised him up to lead Israel out of Egypt, and Rick Doblin has spent 40 years bringing MDMA-AT out of illegality?
The footnote slaps.